



30 May 2017
Subduing the Rebellion: Unmasking Rogue Cells in the Immune System

The immune system is a formidable defense against microbial intruders, identifying and eliminating the threats through an extremely intricate and adaptable network of specialized cells. However, a system of such mind-boggling complexity is also prone to misfire. Due to genetic mutations for example, some of these specialized cells in your immune system might turn rogue: they can no longer differentiate between invaders and your own cells, and thus start attacking healthy tissues. For serious cases, this issue leads to “auto-immune diseases” – such as arthritis, inflammatory bowel disease, type 1 diabetes and many more – which are widespread in the population.
Most of the current treatments against auto-immune diseases require to shut down major parts of the immune system, inhibiting desirable immune responses and, leaving the patient vulnerable to potentially life-threatening bacterial and viral infections. Such a drastic solution is required because until now scientists had yet not fully identified a different mechanism in these rogue cells they could use as a selective target – the known genetical material was common between healthy and rogue cells. But in their publication in Nature Communications, researchers in Okinawa, Japan reported a previously undiscovered role for a known molecule - named “JunB” – and its associated gene: JunB seems to be essential for a specific type of white blood cell to turn toxic.
The scientists were investigating T Helper white blood cells, which coordinate the immune system response by secreting a diverse range of communication signals in the shape of molecules called interleukins. JunB operates in ‘T Helper 17’ cells, a subdivision that specifically promotes the initial immune response against an infection. But sometimes, these T Helper 17 cells turns rogue and become toxic for our guts and joints.
“We have a lot of T Helper 17 cells in our guts” commented Prof. Ishikawa, author of the new study at the Okinawa Institute of Science and Technology Graduate University. “They have three major impacts on our body: first to maintain a healthy gut and second to deal with bacterial and fungi infections. The third is their toxicity leading to auto-immune diseases, which is something we want to avoid.”
The OIST scientists studied the process in which T-Helper 17 cells become toxic. One of the immune system communication molecules – interleukin 23 - is required to “wake up” T-Helper 17 cells during an infection and make it start fighting the invaders. But interleukin 23 is a double-edged sword: it also responsible for sometimes triggering the same T Helper 17 cell to turn rogue. For T helper 17 cells to hear the wake-up call, they need to display interleukin 23 receptors on their surface, which means the corresponding gene – usually switched off – needs to be activated. Finding a way to stop this interleukin 23 receptor gene from being activated is potentially key to shut down the entire process. And this is where JunB, the focus of this research, comes into play.
JunB is a transcription factor, which means it regulates – switches on and/or off - the activity of a gene or a group of genes in the cell’s DNA. OIST researchers from the Immune Signal Unit identified JunB by systematically checking transcription factors in the T Helper 17 cell DNA. For this purpose, they would “knock down” – meaning disrupt the gene DNA sequence – the genes for each transcription factor one by one. For each knocked down gene, they checked if the T Helper 17 cell still display its interleukin-23 receptors. When they knocked down JunB, they realized the T Helper 17 cells were no longer displaying interleukin 23 receptors and were unable to turn rogue. Moreover, mice in which T Helper cells lacked JunB were incapable of developing T Helper 17-related auto-immune diseases.
“It was quite a challenge: we knocked down around three hundred transcription factors, one by one,” said Prof. Ishikawa. “Developing a mice model with a knocked out JunB took us two years!”
Another exciting part of this research is that T Helper 17 cells deprived of JunB are still able to accumulate in the gut and likely to cope and fight infections. Therefore, if scientists design a drug able to target JunB specifically, it should prevent T Helper 17 cells from turning rogue without impacting the immune system as a whole.
In this direction, OIST researchers went onto investigate the exact role of JunB, and this is where it gets a little complex. As a transcription factor, JunB activates another gene - RORγ-t – by helping three other transcription factors bind to the DNA sequence in the chromosome. The product of this gene RORγ-t is a fifth transcription factor which activates the gene Interleukin-23 receptor. Only then the T Helper 17 cell displays interleukin 23 receptors and might turn rogue.
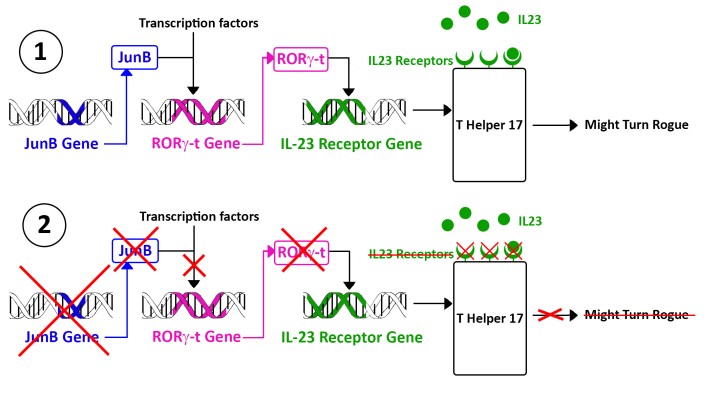
The scientists hope to deepen their knowledge of JunB considering a future treatment against T Helper-17 related auto-immune diseases. “Previously, several transcription factors such as RORγ-t have been found essential for all T Helper 17 cells.” added Prof. Ishikawa. “If we use these molecules as targets for therapy, all T Helper 17 cells will be affected, toxic or not. However, JunB seems to be critical only for toxic T Helper 17 cells: this would allow us to develop a pinpointing, more selective therapy.”
Specialties
Research Unit
Submit a press inquiry









