



15 October 2021
Key protein linked to appetite and obesity in mice

Highlights
- Obesity is an increasingly serious public health issue, but the underlying molecular causes are not fully understood
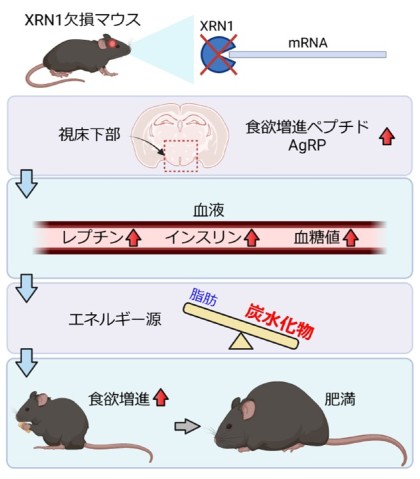
- Now, scientists have found that loss of the protein, XRN1, which is essential for degrading mRNA, from the forebrain in mice leads to extreme appetite and obesity
- Mice without XRN1 are resistant to both leptin, a hormone that suppresses appetite, and insulin, a hormone that regulates the level of glucose in the blood
- Mice without XRN1 accumulate fat in their liver and adipose tissues and cannot use fat effectively as an energy source
- Loss of XRN1 also results in increased activity of a type of neuron in the hypothalamus, which releases a powerful appetite stimulant called AgRP
Main text
Researchers from the Okinawa Institute of Science and Technology Graduate University (OIST) have identified a protein that plays a key role in how the brain regulates appetite and metabolism. Loss of the protein, XRN1, from the forebrain, resulted in obese mice with an insatiable appetite, according to a new study published in the journal, iScience.
Obesity is a growing public health concern, with over 650 million adults worldwide designated as obese. The condition has been linked to many disorders, including cardiovascular disease, type 2 diabetes and cancer.
“Fundamentally, obesity is caused by an imbalance between food intake and energy expenditure,” said Dr. Akiko Yanagiya, a researcher in the Cell Signal Unit at OIST, headed by Professor Tadashi Yamamoto. “But we still understand very little about how appetite or metabolism is regulated by communication between the brain and parts of the body, such as the pancreas, liver and adipose tissues.”
In the study, the scientists created mice that were unable to produce the protein, XRN1, in a subset of neurons in the forebrain. This brain region includes the hypothalamus, an almond-sized structure that releases hormones into the body, helping to regulate body temperature, sleep, thirst and hunger.
At 6-weeks-old, the scientists noticed that the mice without XRN1 in the brain rapidly began to gain weight and became obese by 12 weeks of age. Fat accumulated in the mice’s body, including within adipose tissue and the liver.
When they monitored feeding behavior, the team found that the mice without XRN1 ate almost twice as much each day as the control mice.
“This finding was really surprising,” said Dr. Shohei Takaoka, a former PhD student from the OIST Cell Signal Unit. “When we first knocked out XRN1 in the brain, we didn’t know exactly what we would find, but this drastic increase in appetite was very unexpected.”
To investigate what might be causing the mice to overeat, the scientists measured the blood levels of leptin – a hormone that suppresses hunger. Compared to the controls, the level of leptin in the blood was abnormally high, which would normally stop the mice from feeling hungry. But unlike the control mice, the mice without XRN1 didn’t respond to the presence of leptin – a condition known as leptin resistance.
The scientists also found that 5-week-old mice were resistant to insulin, a hormone that is released by beta cells in the pancreas in response to the high levels of blood glucose that occur after eating. This type of failure in how the body responds to glucose and insulin can ultimately lead to diabetes. As the mice got older, levels of glucose and insulin in the blood rose significantly alongside the increased leptin levels.
“We think that the levels of glucose and insulin rose due to the lack of response to leptin,” explained Dr. Yanagiya. “Leptin resistance meant that the mice kept eating, keeping the level of glucose in the blood high, and therefore increasing insulin in the blood.”
The scientists then checked whether the obesity was also driven by the mice using less energy. They placed each mouse in a special cage that measured how much oxygen the mice used to indirectly work out their metabolic rate.
In the mice aged 6 weeks, the scientists didn’t find an overall difference in energy expenditure. However, they found something very surprising. The mice without XRN1 were mainly using carbohydrates as an energy source, while the control mice were able to switch between burning carbohydrate at night, when they were most active, and fat during the day, when less active.
“For some reason, this means that without XRN1, the mice cannot use fat as a fuel effectively,” said Dr. Yanagiya. “Why this occurs though, we still don’t know.”
Once the mice reached 12 weeks of age, their energy expenditure decreased compared to control mice. But, the scientists believed, this was an effect of obesity, due to the mice being less active, rather than a cause.
“Overall, we think overeating due to leptin resistance was the driving cause behind why these mice became obese,” said Dr. Yanagiya.
To further investigate how loss of XRN1 results in leptin resistance and an increased appetite, the scientists looked at whether the activity of appetite-regulating genes changed within the hypothalamus.
XRN1 plays a crucial role in gene activity, as it is involved in the last step of degrading messenger RNA (mRNA). When a gene is active, DNA is used to make a molecule of mRNA, which can then be used to build a specific protein. Cells have many ways of regulating the activity of genes, one of which is by degrading mRNA more slowly or more quickly, which results in more or less protein being made, respectively.
In the hypothalamus, the scientists found that the mRNA used to make the protein Agouti-related peptide (AgRP) – one of the most potent stimulators of appetite – was elevated in the obese mice, leading to higher amounts of AgRP protein.
“It’s still only speculation, but we think that an increase of this protein, and abnormal activation of the neuron that produces it, might be the cause of leptin resistance in these mice,” said Dr. Yanagiya. “Leptin normally suppresses activity of the AgRP neuron, but if loss of XRN1 results in this neuron remaining highly active, it could override the leptin signal.”
However, the exact mechanism of how loss of XRN1 leads to increased activation of AgRP neurons remains unclear. XRN1 was removed only in a specific subset of neurons in the forebrain, and AgRP neurons were not among them. This suggests that another neuron that did lose XRN1 may be involved and could be signaling incorrectly to the AgRP neurons and keeping them active.
Moving forward, the lab hopes to collaborate with neuroscience research units, in order to pinpoint exactly how XRN1 impacts the activity of neurons in the hypothalamus to regulate appetite.
“Identifying which neurons and proteins in the brain are involved in regulating appetite, and fully determining how resistance to leptin is caused, could eventually lead to a targeted treatment for obesity,” said Dr. Yanagiya.

Article Information
Title: Neuronal XRN1 is required for maintenance of whole-body metabolic homeostasis
Journal: iScience
Authors: Shohei Takaoka, Akiko Yanagiya, Haytham Mohamed Aly Mohamed, Rei Higa, Takaya Abe Ken-ichi Inoue, Akinori Takahashi, Patrick Stoney, Tadashi Yamamoto
Date: 21 September 2021 online
DOI: https://doi.org/10.1016/j.isci.2021.103151

Research Unit
Submit a press inquiry







