



19 March 2021
Reading the book of life

To a biologist who lived a few centuries ago, the biological world may have seemed confusingly diverse. Other than the fact of being alive, what did bacteria or a bonsai tree have in common with a mouse or a mushroom? But due to a burst of biological discoveries in the 19th and 20th century, scientists have now pinpointed one thing that all forms of cellular life here on earth share – a language passed down from our oldest common ancestor. Not one of speech, like English or Japanese, but the language of our genes.
On the surface, this genetic language – encoded in DNA molecules – seems deceptively simple. After all, it’s made up of only four letters, or bases: A, T, C and G. Long sentences of this genetic alphabet make up genes, which act as lines of instruction, commanding the cells in our bodies to pump out RNA and protein. And all the DNA together forms the genome, which plays a pivotal role in how every life form develops and stays alive.
Reading the genome and comparing genetic sequences between organisms were some of the greatest biological and computational challenges of the 20th century. And one of the scientists at the forefront of this effort was Eugene “Gene” Myers, who joined the Okinawa Institute of Science and Technology Graduate University (OIST) as an Adjunct Professor last year.
“I’m a computer scientist by training, and my particular technical interest is in the computer methods needed to solve biological problems,” said Professor Gene Myers.

A pioneer of bioinformatics
Professor Myers is internationally recognized as one of the pioneers of bioinformatics – a relatively young field that uses computers to deal with large and complex biological data, like DNA sequences.
He first dabbled with bioinformatics in the late 1970s as a PhD student at the University of Colorado (“although it was so new then that it didn’t yet have a name,” he noted). But his initial career-defining contribution came in 1990 while working at the University of Arizona, when he developed the core algorithm for BLAST – a software program used to compare and identify sequences of DNA.
Faster and more accurate than previous algorithms, BLAST was a huge success, albeit unnoticed by Professor Myers. “I didn’t even realize that BLAST was getting cited so much until much later on,” he remarked with a wry grin.
With over 89,000 citations and counting, BLAST is one of the ten most highly cited scientific works of all time and is an integral research tool for scientists across the globe.
With his work on BLAST increasing his prominence in the field of bioinformatics, Professor Myers then embarked on his most ambitious and risky project of his career: decoding the human genome using a paired-end shotgun sequencing approach.
At the time, reading the letters, or bases, of DNA could only be done with relatively short pieces, between 100-1000 bases in length. But the human genome is millions of times larger, stretching on for 3.2 billion bases.
One solution to this problem that was advocated by Professor Myers was “paired-end” shotgun sequencing – an improved method of shotgun sequencing more suitable for genomes with areas of DNA that repeat, like the human genome.
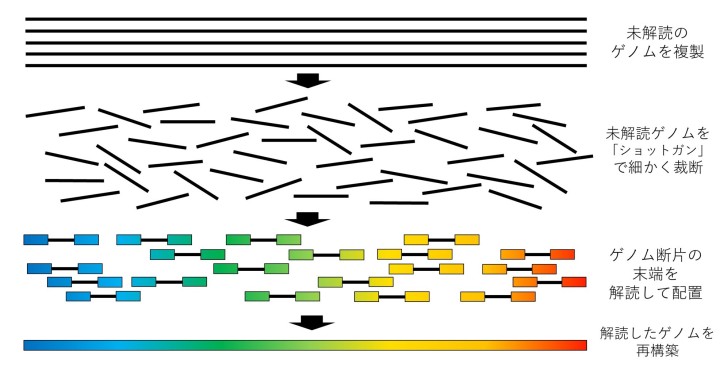
“Shotgun sequencing had been used for smaller genomes, but everyone said it couldn’t be done at the scale of the human genome, especially given the amount of repetitive DNA in the genome. We wrote an article about how we would do it using simulations, but we couldn’t get it published for a year and a half – and only then with an accompanying rebuttal,” he recollected. “No-one would go near this, but I knew I was onto something.”
It wasn’t until 1998 when the private biotech company, Celera Genomics, was formed that Professor Myers got his break. With Celera’s backing, he took a gamble and left academia on a mission to shotgun sequence the human genome.
“There was no guarantee that it would succeed,” he said. “But I was willing to take a chance. And, well, you know the rest of the story – it worked.”
The paired-end shotgun method resulted in the human genome being decoded years ahead of schedule, at a cost of only $100 million – a fraction of the $3 billion needed for the publicly funded Human Genome Project.
But despite his success, Professor Myers remains modest about his accomplishments. “I’d never have expected my career to go this way – I’m very lucky,” he said. “I think what counts in research is to be prepared and take exciting chances when they arrive.”
Constructing perfect genomes
Now back in academia, Professor Myers currently works as a Director of the Max Planck Institute for Molecular Cell Biology and Genetics and the Center for Systems Biology in Dresden, Germany. There, he has shifted his focus to imaging, building microscopes that allow scientists to see the world within cells and organisms to an even higher resolution.
But the latest generation of sequencing technologies, known as long-read sequencing, has tempted him back to constructing genomes – which is central to his research at OIST.
“These new sequencing machines can read much longer fragments of DNA than ever before with higher accuracy,” he enthused. He explained that this makes it easier for scientists to reconstruct new genomes from scratch, without relying on prior information about where genes are located.
Before long-read sequencing, scientists had never been able to construct truly complete genomes. Some regions of the genome, like the telomeres and centromeres at the ends and in the middle of chromosomes were simply too difficult to assemble via shotgun sequencing.
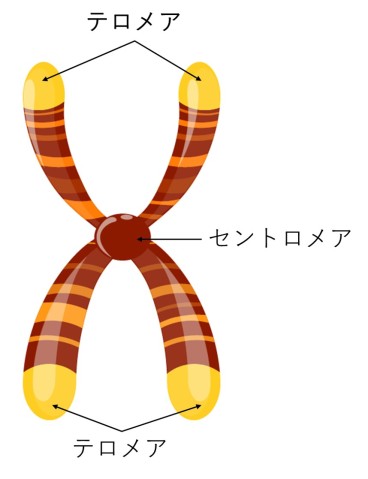
“We had most of the genes, but we didn’t quite know the order of things. But if you’re interested in evolution and want to know how genomes change, then you need the whole genome,” said Professor Myers. “Now, we can take a blood or tissue sample from a living thing, sequence it and reconstruct a genome that is actually better quality than the human genome we’ve had for the last ten years – results that will stand the test of time.”
With new state-of-the-art sequencing technology at his disposal, Professor Myers plans to spend his time at OIST improving the algorithms, to get ever closer to achieving a perfect reconstruction of a genome.
His new unit, Algorithms for Ecological and Evolutionary Genomics, will be a small, focused group with two postdoctoral researchers and two technicians, with the technicians based in the Scientific Computing & Data Analysis Section and the DNA Sequencing Section. They will provide sequencing and bioinformatic services that scientists from all biological fields across the university can benefit from.
For Professor Myers, the interdisciplinary and cooperative nature of bioinformatics is crucial. “I view my work as producing tools that can be used by biologists to enhance their science,” he said. “And one of the ways to make sure that happens is to be involved in collaborative projects.”
To that end, he is collaborating with three units at OIST. One of his projects, with Professor Nori Satoh who leads the Marine Genomics Unit, has an ecological angle. Together, they are working on sequencing the genome of the crown-of-thorns starfish – a coral reef predator that can explode in numbers and devastate large swathes of coral reef. They will also be sequencing the genome of the blue starfish, that lives in the same habitat but doesn’t have these outbreaks, in order to understand the genetics underlying these ecological differences.
Professor Myers has also started collaborating with Professor Evan Economo’s Biodiversity and Biocomplexity Unit. They are using genome sequencing to peer into the evolutionary past of ants and determine how their jaws – or mandibles – have evolved in form and function.
Finally, he is conducting an encyclopedic project with Professor Tom Bourguignon, who leads the Evolutionary Genomics Unit, with a goal of sequencing the genome of every single species of termite.
“With the advent of long-read sequencing, there are a lot of people who now are excited about going out there and sequencing all of nature, and I want to be part of that. That’s the bigger picture,” said Professor Myers.

Although new to OIST, Professor Myers is no stranger to Japan, having lived in Yokohama for three years as a child. He is looking forward to coming to OIST in person, which he hopes to do once travel restrictions are lifted.
“I love Japan – I like the people, culture, and cuisine,” he said. “And I’m impressed by OIST – it’s a very up-and-coming place. I’m excited to be a part of it.”
Specialties
Research Unit
Submit a press inquiry




