



Cellular and Molecular Synaptic Function Unit
Principal Investigator: Tomoyuki Takahashi
Research Theme: Regulatory mechanisms for transmitter release

Abstract
In the neuronal system, dynamic changes of synaptic strength play critical roles in switching functional neuronal circuits. Regarding synaptic strength, compared with postsynaptic mechanisms much less is known for presynaptic mechanisms, primarily because of small nerve terminal structures preventing applications of electrophysiological and imaging techniques. The calyx of Held is a giant glutamatergic nerve terminal visually identified in the mammalian auditory brainstem slices. This fast relay synapse undergoes dramatic developmental changes in its structure, functional properties and molecular compositions during the second postnatal (P) week, when rodents start to hear sound at P10-12. By applying molecular, imaging and patch-clamp techniques to this synapse of developing rodents, we aim at elucidating presynaptic regulatory mechanisms underlying synaptic transmission. Our progress in the fiscal year 2010 is as follows.
- The II-III loop of the a subunit of voltage-gated Ca2+ channels comprises a binding region called synprint site, as it can bind to the exocytic machinery protein SNARE. Given that this binding is Ca2+ concentration-dependent, and that loading of synprint site fragment into presynaptic cells attenuates synaptic responses, it has been hypothesized that this site plays an essential role in exocytic release of neurotransmitter (synprint hypothesis). While screening for proteins, which can bind to the synprint site, Watanabe et al (J Neurosci, 2010) found a direct binding of the synprint with the adaptor protein for clathrin-coated vesicle endocytosis AP-2. In brain lysate, this binding occurred preferentially at low Ca2+ concentration, whereas at high Ca2+ concentration above 100 mM, synaptotagmin (Syt) competed off the AP-2-synprint binding. When the synprint site fragment was directly loaded into the calyx terminal via whole-cell recording pipette, it blocked vesicle endocytosis, assessed by capacitance measurements, without inhibiting exocytosis. This former finding is entirely new and the latter finding is contradictory to the ongoing synprint hypothesis. It is concluded that the voltage-gated Ca2+ channel structure plays essential roles in vesicle endocytosis by binding of its synprint site to AP-2 and Syt in a Ca2+ concentration-dependent manner. Block of these interactions primarily blocks vesicle endocytosis, and will eventually attenuate exocytic release of neurotransmitter because of impaired vesicle recycling and replenishment.
- In the nerve terminal, in response to a presynaptic action potential, a high concentration Ca2+ domain of tens of nanometer in diameter is transiently formed around the site of Ca2+ entry. This Ca2+ nanodomain triggers exocytosis of synaptic vesicles for transmitter release. Yamashita et al (Nat Neurosci, 2010) discovered that this Ca2+ nanodomain plays a priming role for vesicle endocytosis at calyceal presynaptic terminals after hearing onset. At immature terminals of pre-hearing animals, It has been reported that endocytois depends upon Ca2+ outside of the Ca2+ nanodomain exclusively in a calmodulin (CaM)-dependent manner. At post-hearing terminals, however, the CaM-dependent mechanism no longer operated, but vesicle endocytosis after mild and massive exocytosis was still entirely dependent upon Ca2+ within and outside of nanodomain, respectively. These results suggest that both low and high affinity Ca2+ binding proteins are involved in vesicle endocytosis at mature presynaptic terminals. The present study has also clarified the indispensable role of G-proteins in endocytosis. Namely, at mature presynaptic terminals, endocytosis is exclusively GTP-dependent, despite the fact that GTP-independent mechanism partially underlies endocytosis at immature synapses. Essential role of Ca2+ nanodomain in vesicle endocytosis is consistent with our recent finding that Ca2+ channels, via its II-III structure, is involved in vesicle endocytosis (Watanabe et al, 2010, see above), and also with previous reports that the low affinity Ca2+ binding protein synaptotagmin plays dual roles in exocytosis and endocytosis of synaptic vesicles.
- Presynaptic Ca2+ imaging has so far been made only from whole terminal. Nakamura et al (Ms submitted) recorded Ca2+ transients in response to a single presynaptic action potential (AP) from a confocal spot at the calyx of Held presynaptic terminal of developing rats. Hot spots were found along the edge of the terminal facing synaptic cleft, almost ubiquitously at immature calyx (P7-8), but less frequently at more developed calyx of hearing rats (P14-15). Pharmacological mimics of post-hearing Ca2+ transients in pre-hearing calyces using Ca2+ channel blockers revealed that the reduction of functional Ca2+ channel density tightens Ca2+-secretion coupling, as deduced from the effect of the presynaptically loaded slow Ca2+ -binding chelator EGTA on synaptic transmission. Given that AP duration becomes shorter during postnatal development, it is suggested that the developmental reduction of Ca2+ influx underlie establishment of fast synaptic transmission with a tight Ca2+-secretion coupling. These results are also consistent with our previous findings that CaM having relatively low Ca2+ binding affinity operates only at prehearing synapses for Ca2+ channel inactivation (Nakamura et al, 2008) and for vesicle endocytosis (Yamashita et al, see above).
1. Staff
- Dr. Hiroshi Takagi, Group leader
- Dr. Kohgaku Eguchi, Researcher
- Dr. Tetsuya Hori, Researcher
- Dr. Yukihiro Nakamura, Researcher
- Dr. Laurent Guillaud, Researcher
- Dr. Setsuko Nakanishi, Researcher
- Dr. Zacharie Taoufiq, Researcher (part time)
- Mr. Dimitar Dimitrov, Technical staff
- Ms. Kaori Egashira, Research Administrator / Secretary
2. Partner Organizations
- Theme: Regulatory mechanisms for transmitter release
- Type of collaboration: Joint research
- Researchers:
- Dr. Naoto Saitoh, Doshisha University Faculty of Life and Medical Sciences
- Dr. Takafumi Miki, Doshisha University Faculty of Life and Medical Sciences
- Theme: Developmental changes in the presynaptic calcium transient profiles associated with transmitter release
- Type of collaboration: Scientific Collaboration
- Researchers:
- Dr. David DiGregorio, Pasteur Institute
- Theme: Developmental changes in the mechanisms underlying synaptic vesicle endocytosis
- Type of collaboration: Scientific Collaboration
- Researchers:
- Dr. Henrique von Gersdorff, Vollum Institute, Oregon Health & Science University
- Theme: Developmental changes in the presynaptic calcium channels
- Type of collaboration: Scientific Collaboration Research, Division of Cellular Structure
- Researchers:
- Dr. Ryuich Shigemoto, National Institute for Physiological Sciences
- Dr. Ko Matsui, National Institute for Physiological Sciences
3. Activities and Findings
3.1 Requirement of Ca2+ nanodomain in vesicle endocytosis at a mature central synapse and developmental changes in the mechanism underlying Ca2+ -dependent vesicle endocytosis (Yamashita et al, Nature Neuroscience, 2010).
In order to maintain synaptic transmission, synaptic vesicles after exocytosis must be replenished by recycling. Ca2+ can be an ideal reporter to inform the magnitude of exocytosis to endocytic machineries to keep a balance between the number of vesicles and membrane area in the terminal. However, it has been controversioal whether Ca2+ is involved in endocytosis. To address this question we utilized the slow and fast Ca2+ chelators EGTA and BAPTA, respectively, having 200-fold different Ca2+ binding on-rate kinetics. Because of this difference in binding speed, Ca2+ close to the VGCC entry site can be blocked by BAPTA, but not by EGTA, when loaded into the terminal, but not by EGTA. We evoked transmitter release using presynaptic Ca2+ current pulses and monitored vesicle exocytosis and endocytosis using membrane capacitance measurements. Our results indicated that endocytosis evoked by short Ca2+ current pulses can be attenuated by EGTA only at immature period before hearing, whereas BAPTA blocks all types of endocytosis even after hearing onset (Fig 1).
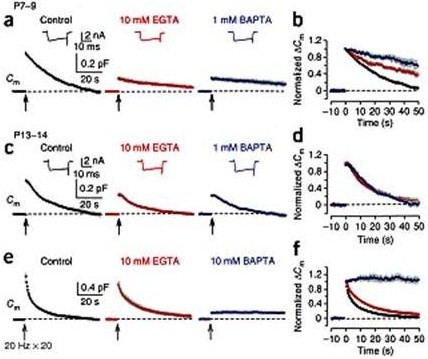
Figure 1 Different effects of EGTA and BAPTA on vesicle endocytosis at the developing calyces of Held. (a) At P7–9 calyceal terminals, presynaptic Ca2+ currents (ICa, upper insets) and membrane capacitance (Cm) changes were evoked by a 20 ms depolarizing command pulse (from –80 to +10 mV, the onset indicated by an arrow), in the presence of 0.5 mM EGTA (control, black), 10 mM EGTA (red) or 1 mM BAPTA (blue) in the whole cell patch pipette. Average Cm traces from 6–8 presynaptic terminals and representative ICa traces are shown. Gray shadings indicate s.e.m. in this (b, d, f) and following figures (Figs. 2, 3). (b) The Cm traces (in a) normalized at the peak. (c, d) The same stimulation protocol as in a was applied to P13–14 calyces. Averaged Cm traces were obtained from 5–9 calyceal terminals. (e) Cm changes induced by a train of 20 depolarizing pulses (20 ms duration) at 20 Hz in the presence of 0.5 mM EGTA (control, black, n = 9), 10 mM EGTA (red, n = 8) or 10 mM BAPTA (blue, n = 5) loaded into P13–14 calyces. EGTA (10 mM) had no significant effect on exocytic DCm (1.07 ± 0.10 pF, P = 0.098). (f) The Cm traces (in e) normalized at the peak. From Yamashita et al. (2010) Nat Naurosci with a permission.
These results suggest that endocytosis requires Ca2+ throughout development, and that the Ca2+ domain in immediate vicinity of VGCC (Ca2+ nanodomain) plays an essential role in reporting the magnitude of exocytosis to endocytic machineries.
During massive vesicle exocytosis, endocytic rate increases by several to 10-fold to compensate for the loss of vesicles. Vesicle endocytosis following massive exocytosis was Ca2+-dependent, being attenuated by EGTA and abolishable with BAPTA (Fig 2a, b, c).
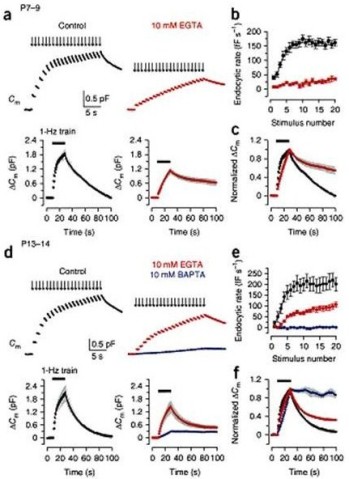
Figure 2 Ca2+-dependence of fast and slow endocytosis elicited by repetitive stimulation at the calyx of Held. (a) Top panels, Cm changes induced by 20 ms depolarizing pulses (arrows indicate onsets) repeated at 1 Hz for 20 s at P7–9 calyceal terminals pre-loaded with EGTA at 0.5 mM (black, control) or 10 mM (red). Bottom panels, averaged Cm records, shown at a longer time scale, from P7–9 calyceal terminals with 0.5 mM EGTA (n = 7) or 10 mM EGTA (n = 5). Black bars indicate the period of stimulation in this and Fig. 3. (b) Endocytic rate during the stimulus train (fF s–1, ordinate), estimated from linear regression line fit to the Cm decay 0.45–1 s after each pulse versus stimulus number (abscissa) in P7–9 calyces with 0.5 mM EGTA (black, n = 16) or 10 mM EGTA (red, n = 6). (c) The Cm traces (shown in the bottom panels of a) normalized at the peak (superimposed). (d–f) The same experimental protocol as in a–c was applied to P13–14 calyces (n = 5–9). Data from terminals loaded with 10 mM BAPTA (blue) are also shown. From Yamashita et al. (2010) Nat Naurosci with a permission.
Direct injection of inhibitor peptides into presynaptic terminals of developing rats revealed that this type of exocytosis was calmodulin (CaM)- and calcineurin (CaN)-dependent before hearing (Fig 3a, b, c). Surprisingly, however, after hearing onset, the CaM/CaN dependent endocytosis became inoperative (Fig 3, d, e, f), despite the fact that its Ca2+-dependence persists (Fig2d, e) and the kinetics and magnitude of endocytosis in the same stimulation protocol remained similar (Fig 3).
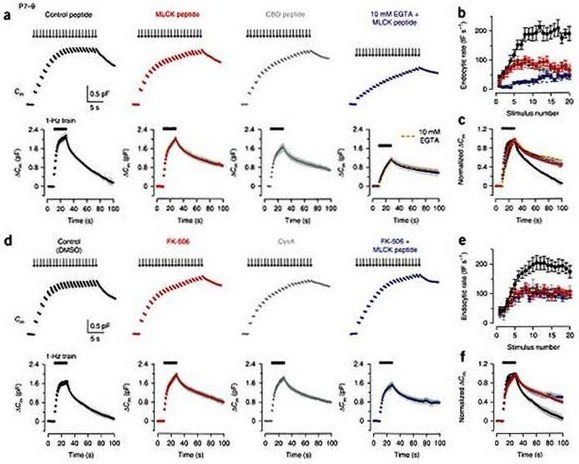
Figure 3 Effects of inhibitors for CaM and CaN in fast and slow endocytosis at P7–9 calyces. (a) Top panels, Cm changes induced by a stimulus train (20 pulses of 20 ms depolarization at 1 Hz) in the terminal preloaded with control peptide (0.2 mM, black, P8), MLCK peptide (0.2 mM, red, P8), CBD peptide (20 mM, dark gray, P8) or MLCK peptide (0.2 mM) together with 10 mM EGTA (blue, P9). Bottom panels, averaged Cm records, shown at a slow time scale, from P7–9 calyceal terminals with these peptides (n = 5 for each). After normalizing the peak amplitude, data with 10 mM EGTA (Fig. 2a) was superimposed (dashed red line) on the data with 10 mM EGTA and MLCK peptide (blue) on the right bottom panel. (b) Summary data for the Cm decay rate (fF s–1, ordinate) measured after each pulse during the 1 Hz train versus stimulus number (abscissa) in the presence of control peptide (black, n = 6), MLCK peptide (red, n = 6), CBD peptide (dark gray, n = 5) or MLCK peptide together with 10 mM EGTA (blue, n = 6). Dashed line indicates data from 10 mM EGTA-loaded terminals (Fig. 2a). (c) The averaged Cm traces, shown in the bottom of a, normalized at the peak (superimposed). (d–f) Similar to a–c, but data from DMSO (0.1%)-loaded control terminals (black) or calyceal terminals loaded with FK-506 (0.1 mM, with 0.1% DMSO, red), cyclosporin A (CysA, 1 mM, with 0.1% DMSO, dark gray) or FK-506 (0.1 mM, with 0.1% DMSO) together with MLCK peptide (0.2 mM, blue), in P7–9 rats. Sample records (top panels of d) were derived from different calyceal terminals in P8 rats. Averaged Cm records (bottom panels of d) were obtained from 6–11 terminals. From Yamashita et al. (2010) Nat Naurosci with a permission.
3.2 Retrograde upregulatory mechanism for synaptic vesicle endocytosis (Eguchi et al, in preparation)
In addition to Ca2+, we have found a retrograde feedback mechanism for reporting the magnitude of vesicle exocytosis to endocytosis. While testing the effects of protein kinase inhibitors by directly injecting them into the calyx of Held presynaptic terminal, we found that protein kinase G (PKG) inhibitors attenuates vesicle endocytosis by slowing the rate of membrane capacitance recovery after exocytosis. This effect of PKG inhibitors could be mimicked an occluded by a bath-applied NO scavenger, and furthermore by the NMDA receptor blocker APV. These results suggest that glutamate released from vesicles by exocytosis activates postsynaptic NMDA receptors and causes a Ca2+ influx, which promotes NO synthesis and NO release from postsynaptic cells, thereby activating PKG in presynaptic terminals in a retrograde manner. We then tested the downstream cascade of PKG toward endocytic mechanism and found that a PIP2 inhibitor mimics and occlude the inhibitory effect of PKG inhibitor on vesicle endocytosis. Furthermore, PIP2 loaded into the calyx terminal blocked the inhibitory effect of the PKG inhibitor, and a PKG inhibitor or NO scavenger reduced the intensity of PIP2 immunoreactivity at the calyx of Held terminal. These results suggest that PIP2 resides downstream of PKG and mediates the message of glutamate to endocytic machinery.
3.3 Synaptic vesicle filling with neurotransmitter as a rate-limiting step for vesicle reuse (Hori et al, in preparation)
After exocytic release of transmitter, synaptic vesicles are retrieved by endocytosis and recycled to be reused. Vesicle recycling rates have been extensively studied using vesicle marking dyes and revealed fast and slow pathways, with their time constants ranging from subseconds to tens of seconds. It has been postulated that the fast and slow recycling pathways operate in parallel, and fast pathway is necessary for fast vesicle reuse, particularly at nerve terminals with small reserve pool of synaptic vesicles. Given that the mean amplitude of miniature, or single vesicle, synaptic currents is unchanged even after prolonged high frequency stimulation, it has been presumed that the vesicle refilling rate is fast enough and not limiting the rate of vesicle reuse. However, glutamate uptake speed into isolated synaptosome (>10 min) is too slow to account for the time for synaptic currents to recover from depression by (~1 min) prolonged high frequency stimulation. To date, at synapses, no direct measurement has been made for the rate of vesicle refilling with neurotransmitter. We have challenged this problem at the calyx of Held terminal. To determine the vesicle refilling rate, we first depleted vesicular glutamate by washing out intra-terminal cytosolic glutamate (Ishikawa et al, 2001) and replace it by membrane impermeable caged MNI-glutamate. We monitored the EPSC amplitude as an index of vesicle filling. As glutamate was washed out by patch pipette solution, EPSCs diminished to a low level. We then apply a short UV pulse to uncage MNI-glutamate, and measure the rate at which the EPSC amplitude increases. Throughout the procedure, the magnitude of exocytic membrane capacitance change remained the same, indicating that constant number of vesicles undergo exocytosis in response to stimulation. Our results suggest that the filling process of synaptic vesicles with glutamate can be a limiting step for vesicle reuse.
4. Publications
4.1 Journals (OIST researchers are underlined)
- Watanabe H., Yamashita T., Saitoh N., Kiyonaka S., Iwamatsu A., Campbell K.P., Mori Y. & Takahashi T. (2010). Involvement of Ca2+ channel synprint site in synaptic vesicle endocytosis. J Neurosci 30, 655-660.
- Yamashita T., Eguchi K., Saitoh N., von Gersdorff H. & Takahashi T. (2010) Nanodomain Ca2+ is essential for vesicle endocytosis at a mature central synapse. Nature Neuroscience 13, 838-844.
4.2 Books and other one-time publications
- Patch clamp recording method in slices for studying presynaptic machanisms. Tomoyuki Takahashi, Tetsuya Hori, Yukihiro Nakamura & Takayuki Yamashita, Springer, in press
4.3 Oral and Poster Presentations
- Hori T. & Takahashi T. Neurotransmitter refilling rate of synaptic vesicles measured in the central presynaptic terminal, The 87th Annual Meeting of the Physiological Society of Japan, Morioka, Iwate, Japan, May 19-21, 2010
- Eguchi K. & Takahashi T. Protein phosphorylation regulates vesicle exo/endocytosis at the calyx of Held synapses, Neuro2010, Kobe, Japan, September 2-4, 2010
- Eguchi K. & Takahashi T. The regulatory mechanisms of protein kinase G-dependent vesicle endocytosis at the calyx of Held synapses, National Institute for Physiological Science Meeting 2010, Okazaki, Aichi, Japan, September 30- October 1, 2010
- Takahashi S., Nakamura Y., Inamura K., Miyane A. Selective involvement of the ether-à-go-go K+ channel BEC1/KCNH3 in cognitive function , 40th annual meeting Neuroscience 2010, San Diego, CA, November 13-17, 2010
5. Intellectual Property Rights and Other Specific Achievements
Nothing to be reported.
6. Meetings and Events
6.1 Seminar
Title: Imaging mass spectrometry and its data analysis
- Date: January 7, 2010
- Venue: Lab 1
- Speakers: Mitsutoshi Setou, Hamamatsu University School of Medicine