



05 April 2017
Fish Eyes to Help Understand Human Inherited Blindness

Newborns babies can be at risk of congenital blindness, presenting sight defects due to lesions or to genetic mutations in their genome. Among the latter, Leber Congenital Amaurosis – or LCA - is one of the most widespread causes of child blindness and accounts for nearly 5% of vision impairments overall. The syndrome can be genetically transmitted to a child when both parents possess at least one dysfunctional copy of a gene involved in eye development. However, the molecular mechanism behind the disease remains unclear. Now OIST researchers in the Developmental Neurobiology Unit have exposed a similar syndrome in zebrafish, which are an excellent model for studying human diseases. From this research published in Scientific Reports, scientists aim to unravel the causes behind the disease in zebrafish and therefore provide new leads for a treatment for human LCA.
LCA affects the retina, the thin layer of tissue at the back of the eye that detects light as well as differentiates colors and communicates the information to the brain via the optic nerve. A healthy retina usually features light-sensitive cells – photoreceptors - called cones and rods. Cones are specialized in bright environment and detect colors while rods are used in dim light but are monochrome, which is why we see in black and white at night. A person with LCA will display deformed or absent cones and rods, thus preventing the detection of light. A total of 24 genes including a gene called Aipl1 – standing for aryl hydrocarbon receptor interacting protein like 1 - have been linked to LCA in humans and mice. The illness occurs when a DNA mutation within one of the genes affects the normal ocular development or induces photoreceptor – the cones and rods - degeneration.
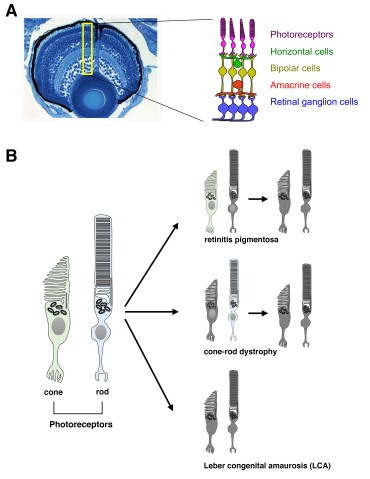
OIST scientists selected the zebrafish as an animal model because its retina is rich in cones and its visual acuity can be measured with a simple device. The researchers studied a genetically mutated zebrafish embryo that did not react to visual stimuli. They discovered that zebrafish DNA contains two Aipl1 genes, namely Aipl1a and Aipl1b, which are respectively active in rods and cones. The mutant - called gold rush (gosh) - presents a genetic mutation in the Aipl1b DNA sequence, losing Aipl1 activity in cone photoreceptors. Consequently, the cone photoreceptors showed a deformed morphology and sustained degeneration. Rods however were not affected, suggesting the degeneration is cone-specific.
Probing further, the authors of the study also revealed that Aipl1 is critical for the stability of two enzymes – the cGMP-phosphodiesterase 6 and the guanylate cyclase - which mediate phototransduction, the process of converting light into an electrical signal. Without these enzymes, the zebrafish is unable to react to light stimulus as the information is stopped in photoreceptors and fails to initiate the transmission of visual information into the brain through the optic nerve.
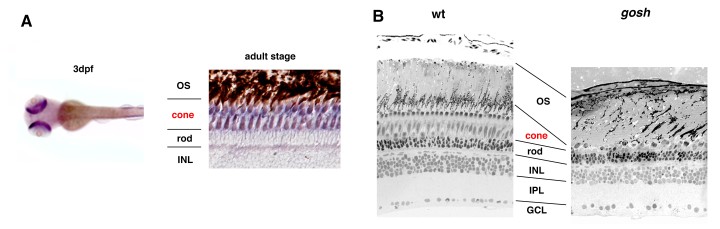
The research indicates that Aipl1b gene is important for visual functions and maintenance of cone photoreceptors in the zebrafish. Without it, cones do not detect light stimuli and degenerate during development, which are clues for treating the illness in humans. Dr. Maria Iribarne, first author of this study, commented: “The gosh mutant is a good model for understanding the molecular and cellular mechanism of cone cell death and the pathological process of human LCA. Hopefully, this new knowledge will help to find a future cure for patients who suffer such a devastating disease as LCA”.

Specialties
Research Unit
Submit a press inquiry







